科研讲堂|一文get基因编辑鼠基因型鉴定
近些年越来越多的基因编辑鼠被引入到各个实验室,很多同学刚入学就被师兄师姐带着养鼠、鉴定鼠等等。今天小编就带着大家一起学习下基因编辑鼠基因型鉴定吧~
基因编辑鼠发展史

图1.基因编辑鼠发展史
Cre/LoxP系统
Cre/LoxP是做基因修饰鼠最常用的工具, 如图2所示,含有Cre(催化两个位点间的基因序列发声环化重组的一种酶)的鼠与含有LoxP(能被Cre特异识别的一段34个碱基的DNA序列)的鼠交配,产生既含有Cre又有LoxP的鼠,在Cre重组酶的催化下,两个LoxP位点的序列发生环化重组删除,从而达到目的基因编辑,通过与组织特异性Cre交配产生特定组织发生基因编辑的条件性基因编辑鼠。

图2.Cre/LoxP系统简单原理图。
CRISPR/Cas9
CRISPR (clustered regularly interspaced short palindromic repeats)是一种原核生物利用RNA引导的DNA核酸酶Cas9对外源的噬菌体或病毒核酸进行基因沉默的获得性免疫系统。如图3所示,在sgRNA的引导下,Cas9核酸酶到达目的基因位点,从而快捷实现对靶基因编辑。
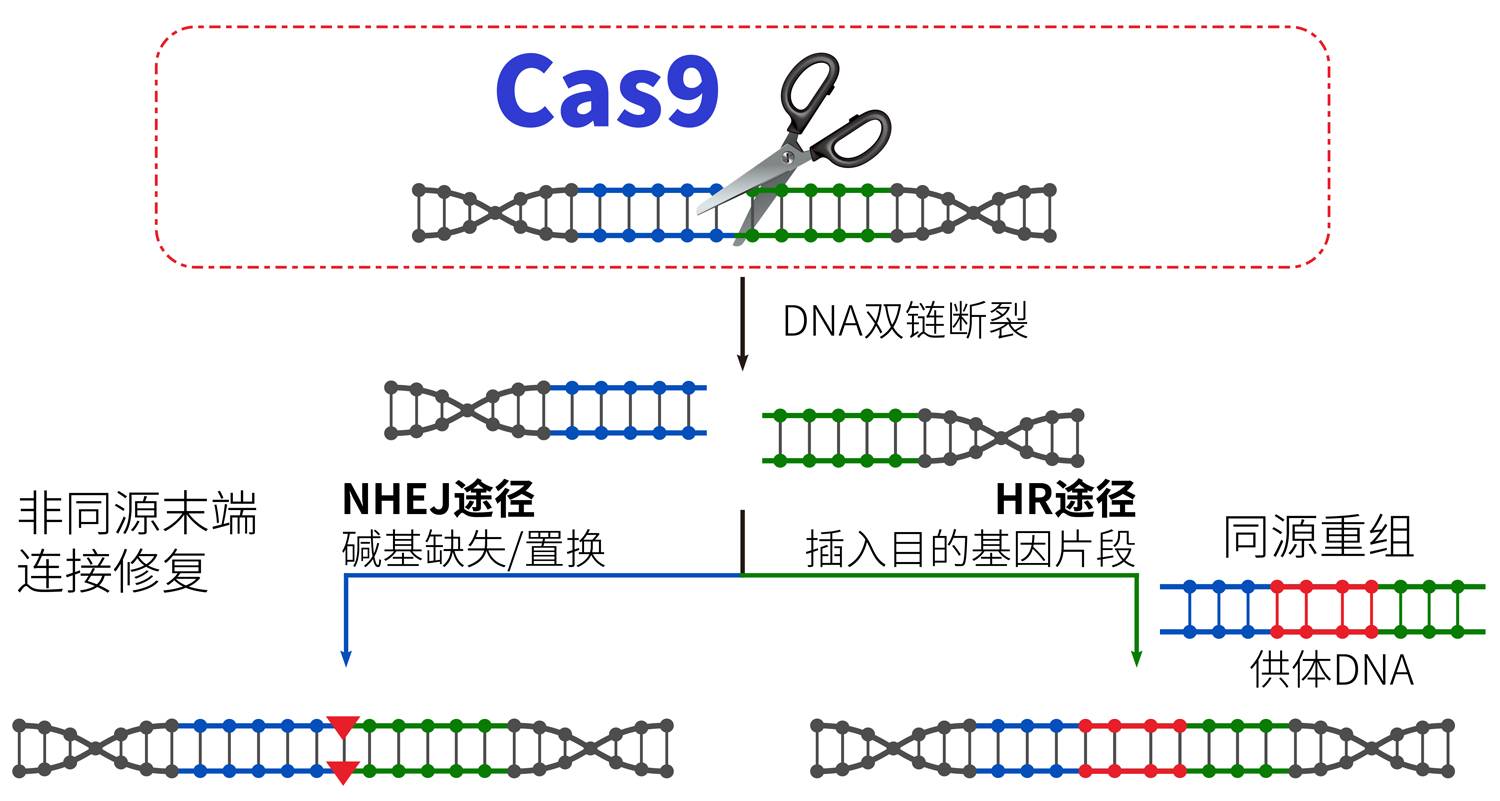
图3.CRISPR/Cas9简单原理图。
基因编辑鼠类型
基因编辑鼠包括:转基因(transgenic)、基因敲入(knock in)、基因敲除(knock out)和基因点突变(point mutation)鼠。
基因编辑鼠基因型鉴定
辛辛苦苦繁殖的鼠,哪些鼠实现了基因编辑?是杂合子(同一位点上的两个等位基因不相同的基因型个体)还是纯合子(同一位点上的两个等位基因相同的基因型个体)这就需要我们通过基因型鉴定来知道啦。常见的基因型鉴定方法包括直接PCR、RT-PCR和测序等。今天主要介绍最常用的直接PCR鉴定法。
基本原理
提取鼠的基因组DNA,通过PCR扩增DNA,利用DNA电泳根据条带大小和条带数量差异区分是否实现了基因编辑。
实验分组
WT Control:用野生型鼠组织或DNA作为对照,协助判断基因型及PCR体系是否合适。
Blank Control:不含有任何组织/DNA的对照组,用来质控PCR体系是否发生污染。
Internal Positive Control: 内部对照,引物通常扩增与目的基因无关的基因组DNA区域,用来判断 PCR 体系和模板质量是否合适。
*碧云天鼠尾鉴定试剂盒里提供了GAPDH的引物,可以作为Internal Positive Control。
Sample:目标小鼠组织或DNA,鉴定基因编辑是否成功。
基因组DNA提取
目前基因组DNA的提取主要有以下两种方法:
1. 碱裂解法:通过一定浓度的碱性溶液消化组织,释放DNA。
优点:成本低。
缺点:各实验室配方各异,提取效果不一,且试剂具有腐蚀危险性。
2. 酶消化法:通过酶(如蛋白酶K)消化组织,释放DNA。
优点:操作简单、效果稳定、较安全。
缺点:成本略高。
小编有话说:
碧云天生产的鼠尾基因型快速鉴定试剂盒(D7283)中使用的是经优化改造的混合酶进行组织消化,仅需20分钟就可完成DNA提取~
此外,碧云天还有不需要DNA提取,直接用血样进行PCR鉴定的试剂盒——一步法鼠基因型鉴定试剂盒(D7284)供大家选择。
常见问题:
Q:组织在孵育后未完全消化。
A:有些组织很难完全消化。碧云天生产的直接PCR试剂盒不要求组织完全消化,部分消化提取的DNA通常也足够满足PCR检测。
PCR扩增
即利用DNA聚合酶,通过PCR反应扩增提取出的目的DNA片段。
小编有话说:
碧云天鼠尾基因型快速鉴定试剂盒(D7283)采用Easy-Load™ PCR Master Mix (Green, 2X)(D7255)进行PCR。该产品稳定性高,经测试,反复冻融15次后对PCR的扩增效果无显著影响。反复冻融15次前后,使用不同引物扩增100-1500bp间不同长度目的片段的效果如图2所示。
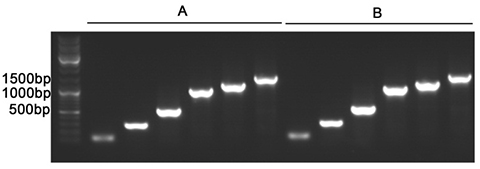
图4. Easy-Load™ PCR Master Mix (Green, 2X)反复冻融15次前后扩增不同长度产物的效果图。A. 反复冻融前本产品进行PCR的扩增结果。B.反复冻融15次后的本产品进行PCR的扩增结果。除本产品是否反复冻融15次外,A和B的反应体系完全相同,电泳的上样量也相同。
琼脂糖凝胶电泳
PCR反应结束后直接上样进行琼脂糖凝胶电泳,根据条带大小和位置进行判断野生型、杂合子和纯合子。
结果解读
点突变鼠只能通过测序的方法进行鉴定,这里只对基因删除及基因插入为目的的基因编辑鼠PCR鉴定结果进行解读。
全身性敲除(KO)
如图5所示,在目的基因上下游分别设计一条引物进行PCR,发生敲除成功的鼠由于片段缺失,所以PCR产物比野生型小,杂合子是只有一条染色体发生了基因片段缺失,另外一条染色体正常,故会出现野生型和敲除两个条带。

图5.全身性敲除示意图。
条件性敲除(CKO)
如图6所示,只含有LoxP的Flox杂合子鼠一条染色体正常,另外一条染色体多出两段LoxP序列,所以会在野生型条带上面出现一条分子量稍微大的Flox条带;当Flox杂合子鼠与Cre鼠交配后,含有LoxP位点的染色体会在Cre酶的作用下发生目的基因环化删除,故会在野生型条带的下方出现一条分子量小的敲除条带;纯合子敲除鼠的两个染色体都发生了敲除,故只有一条分子量小的敲除条带出现。
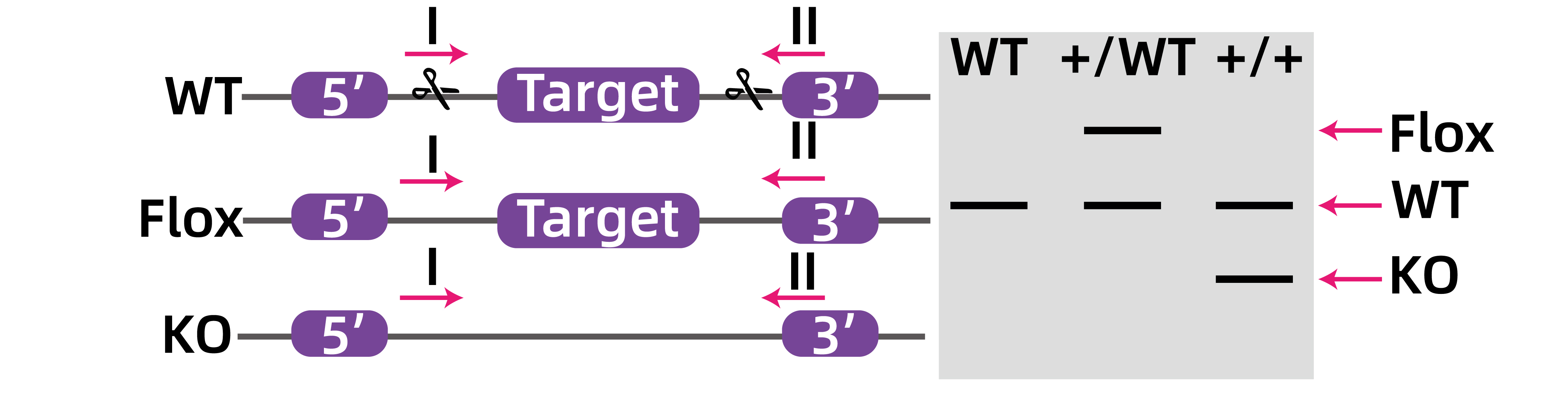
图6.条件性敲除示意图。
转基因/敲入(KI)
如图7所示,对于插入的基因,一般会设计两对引物(I、II:插入基因上下游;III、IV:插入序列上与下游同源臂上),根据条带大小的差异区分是否成功插入目的基因,根据III、IV引物设计的位点,目的条带可能大于或小于野生型条带,此处以小于野生型条带为例。
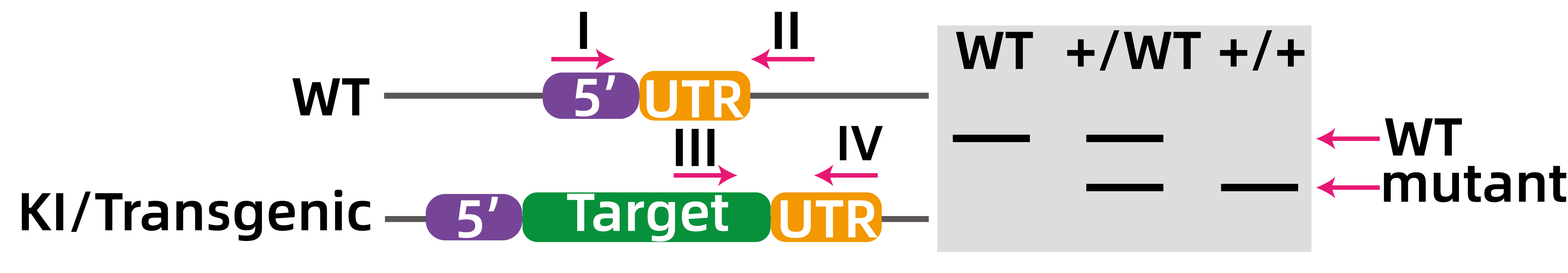
图7.转基因/敲入示意图。
常见问题:
Q:出现三个条带。
A:聚合酶效率太高,杂合子两个条带发生了反应,从而出现了第三个条带。所以,三个条带的结果可以直接当做杂合子,不影响结果判读。如果要求严格,可以换反应效率低的聚合酶。
Q:PCR产物少或没有目的条带。
A:(滑动下方看全文)
a. 组织提取物中的污染物抑制了PCR反应。为检测抑制物,可用等体积混合的DNA Extraction Solution和Stop Solution,同时用DNA对照或已知数量的模板(100-500 copies)进行PCR反应。
b. 组织消化不够充分。可适当延长55℃消化时间或适当增加Enzyme Mix的使用量。
c. Enzyme Mix未被完全灭活。适当延长消化产物在95℃的孵育时间。
d. 引物设计不佳是PCR过程中最常见的问题。请选择适当的引物设计软件进行引物设计,注意引物的GC含量、二级结构、二聚体、退火温度、长度、特异性等方面的问题。在加入酶切位点等的引物中,一定要注意加入酶切位点等后整条引物的GC含量、二级结构、二聚体、退火温度、长度、特异性等方面的问题。在原有引物效果不佳的情况并且阳性对照引物可以正常工作的情况下,可以考虑更换引物。
e. 待扩增片段GC含量偏高。GC含量较高的情况下PCR会变得相对比较困难,此时可以使用适合扩增高GC含量DNA片段的GC-rich buffer,并相应地根据GC-rich buffer的要求或说明调整PCR反应参数的设置。
f. 长片段扩增。尽管Taq DNA polymerase可以扩增最长达8kb的DNA片段,但大多数时候比较适合扩增2-3kb以下的片段,更长片段的扩增推荐使用其它更适合长片段扩增的DNA聚合酶。
g. PCR反应设置时在室温进行容易导致非特异性条件。推荐在冰浴上设置PCR反应。
h. 由于引物存在一定的二级结构或存在一定的引物二聚体,或引物偏短,导致退火效果不佳。此时可以采用Touch down等方法进行退火,通常采用从65℃逐步缓慢降温到55℃或50℃的方法,使退火更加充分。
i. 退火温度不佳,需要优化。如果有温度梯度PCR仪,则可以设置退火的温度梯度,摸索退火的最佳温度。如果没有温度梯度PCR仪,则可以通过多次PCR反应摸索最佳的退火温度。
j. 延伸时间不足。可按照每1kb片段延伸1分钟进行设置,对于较难扩增的片段可以设置为每1kb片段延伸1.5-2分钟。
k. 待扩增片段GC含量较高或长度较长,变性不够充分。可以调节起始变性条件至95℃ 1min甚至95℃ 2-4min。
l. 在不同PCR仪上进行PCR反应,避免有时PCR仪出现问题。
m. 循环数不足,适当延长PCR的循环数。通常循环数最高不必超过40,常用的循环数范围为25-35。
n. 模板含量太低,适当加大模板量,或采用巢式PCR(nested PCR)或二次PCR。巢式PCR即为在原先设计的PCR引物内侧再设计一对PCR引物,然后对第一次PCR产物进行稀释后再进行一次PCR扩增,这样一方面可以起到扩增作用,同时也可以从第一次PCR产物中扩增出特异性条带。二次PCR则为比较简单地用原有引物对第一次PCR产物进行稀释后再进行一次PCR扩增,可以起到扩增作用,但不能去除非特异性条带。
o. 注意设置适当的阳性对照和阴性对照通常会有很大帮助。
相关产品信息
| 产品编号 | 产品名称 | 产品包装 |
| D7284S | 一步法鼠基因型鉴定试剂盒 | 100次 |
| D7284M | 一步法鼠基因型鉴定试剂盒 | 500次 |
| D7283M | 鼠尾基因型快速鉴定试剂盒 | 500次 |
| D7283S | 鼠尾基因型快速鉴定试剂盒 | 100次 |